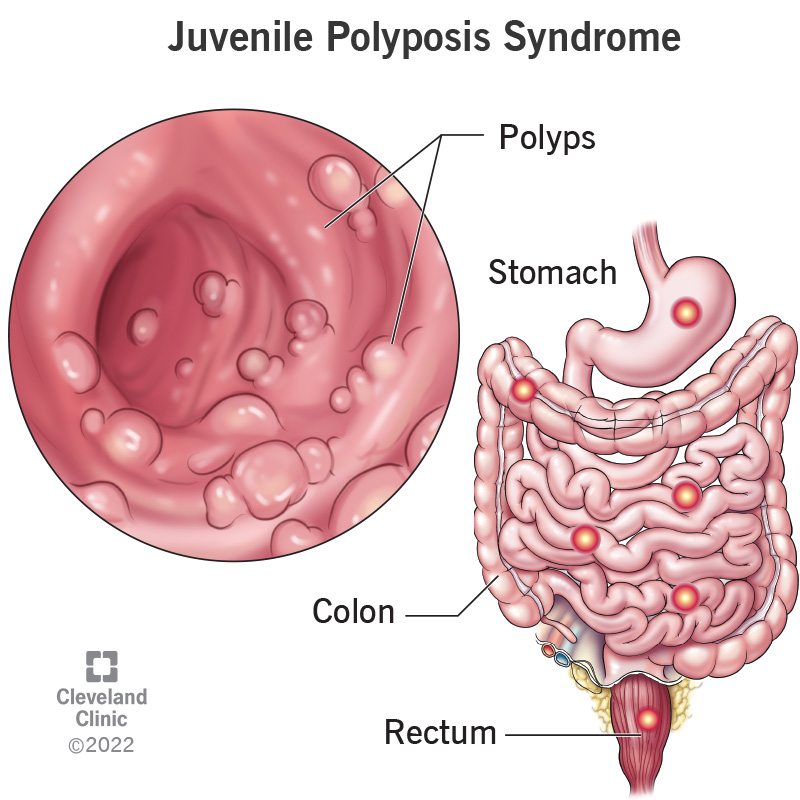
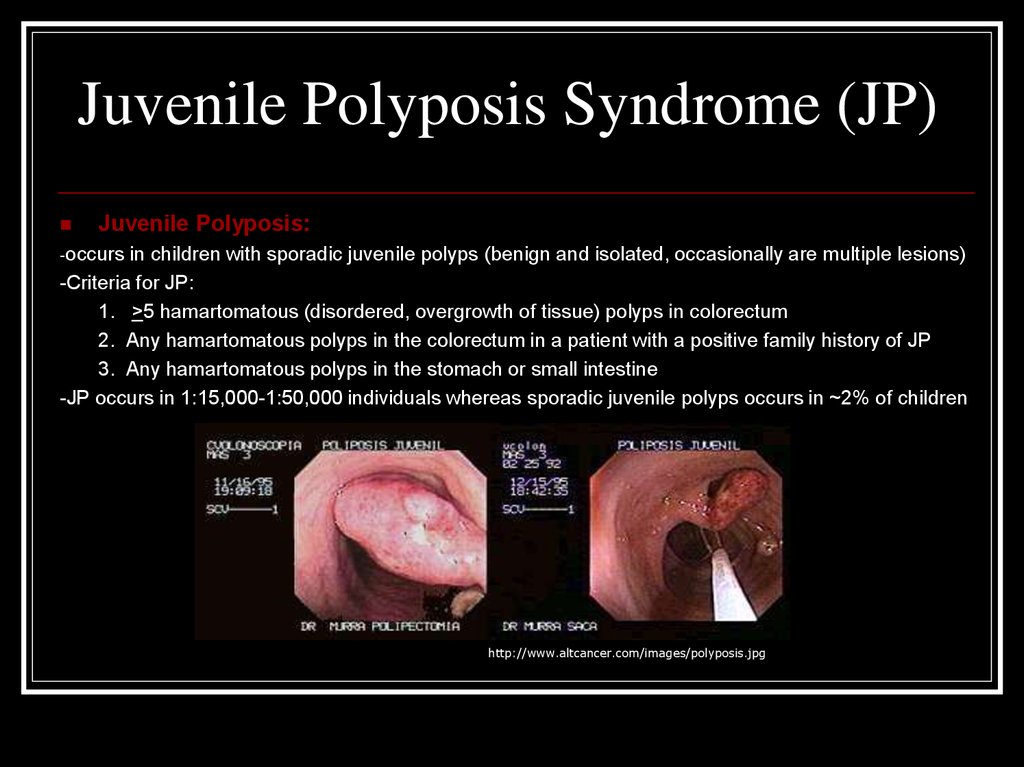
El síndrome de poliposis juvenil es una enfermedad poco frecuente de causa genética y herencia autosómica dominante que se caracteriza por la aparición de pólipos hamartomatosos en el intestino durante el periodo infantil o juvenil, aunque también pueden desarrollarse en la edad adulta.[1] [2] [3]
Descripción
Se estima que se presenta un caso por cada 50 000 nacidos vivos. La edad típica de inicio de las manifestaciones es la infancia o adolescencia. Se han descrito diferentes variantes dependiendo de la gravedad de la afección y si los pólipos se localizan en intestino delgado, intestino grueso o tracto digestivo superior. Las manifestaciones principales son sangrado rectal, anemia y dolor abdominal, en algunos casos puede existir retraso del crecimiento. La enfermedad es hereditaria según un patrón autosómico dominante, se han detectado mutaciones asociadas en los genes SMAD4 (18q21.1) y BMPR1A (10q22.3).[3] El diagnóstico diferencial se realiza con otros cuadros de manifestaciones similares, entre ellos el síndrome de Bannayan–Riley–Ruvalcaba, el síndrome de Cowden, la poliposis adenomatosa familiar y el síndrome de Peutz-Jeghers. Uno de los riesgos principales al que se enfrentan los pacientes afectados por este síndrome es la alta probabilidad de desarrollar cáncer gastrointestinal o cáncer de páncreas, por lo que deben someterse a revisiones periódicas. En algunos casos se recomienda la colectomía quirúrgica preventiva.[1]
Referencias